Developing and testing docking methods in model systems
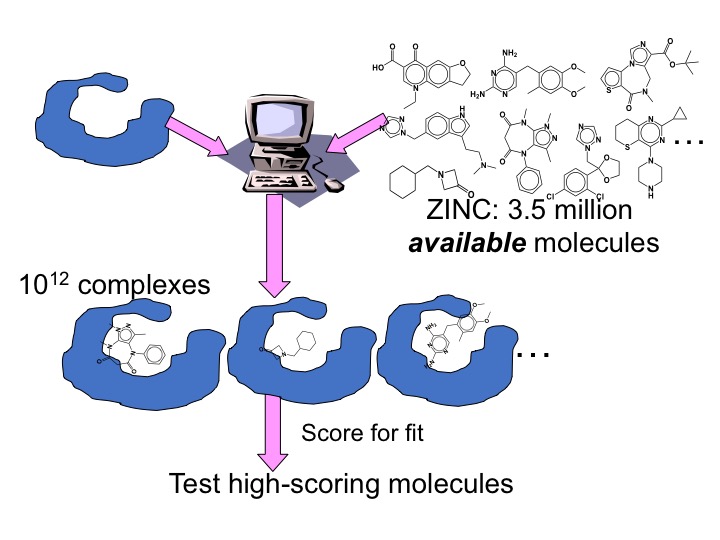 Molecular docking screens libraries of molecules for physical fit to a protein site; millions of molecules are sampled, each in around 100,000 orientations and conformations (about 1013 complexes total). Each configuration of each molecule is scored for complementarity, and high-ranking ones are tested for binding and efficacy. Especially in the last decade, docking has revealed interesting new ligands for bio-relevant targets.
Molecular docking screens libraries of molecules for physical fit to a protein site; millions of molecules are sampled, each in around 100,000 orientations and conformations (about 1013 complexes total). Each configuration of each molecule is scored for complementarity, and high-ranking ones are tested for binding and efficacy. Especially in the last decade, docking has revealed interesting new ligands for bio-relevant targets.
Not withstanding these successes, the technique retains great liabilities: under-sampling conformations, crudely approximating interaction energies, struggling to model bridging and solvating water, among other terms. It cannot predict ligand affinities, nor even rank-order diverse ligands. Worse still, we rarely understand why docking fails when it does, frustrating optimization.
There are two strategies in physics and biology to treat complicated problems with entangled terms: methods development from first principles, or simplifying the problem to the point of tractability. We have adopted a dual approach, engineering radically simplified protein sites to isolate specific terms in docking and test them experimentally, before extending new methods to biologically relevant targets, often GPCRs.
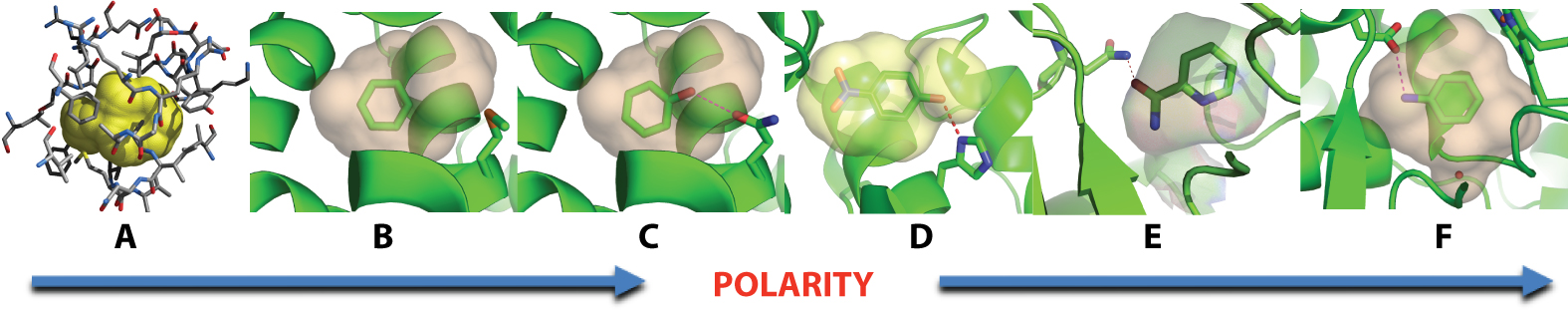
Six protein cavities sites have been engineered for docking. Each is small (150 to 200 Å3), buried from solvent, and dominated by a single term: from a fully apolar cavity, to the same cavity with a single hydrogen-bond acceptor, to an anionic cavity dominated by a single aspartate, and intermediates between. Against each cavity we screen a library of over 500,000 molecules, testing predicted ligands for affinity and for geometric fidelity by crystallography. The simplicity of these sites allows us to isolate and test particular docking terms. Uniquely, in these sites failed predictions can be more illuminating than successes. Recent papers include:
- M Fischer et al. Incorporation of protein flexibility & conformational energy penalties in docking screens to improve ligand discovery. Nature Chemistry 6, 575-83 (2014). PMC4539595.
- N London, et al. Covalent Docking of Large Libraries for the Discovery of Chemical Probes. Nature Chem. Biol. 10, 1066-72 (2014). PMC4232467.
- M Merski et al. Homologous ligands accommodated by discrete conformations of a buried cavity. PNAS 112, 5039-44 (2015). PMC4413287.
- TE Balius et al. Testing inhomogeneous solvation theory in structure-based ligand discovery. PNAS 114, E6839-E684 (2017). PMC5565424.
As these data may be useful to the community, we provide below sets of molecules both observed to and not observed to bind to the model cavity sites.
 |
|
 |
|
 |
|
 |
|
 |
**Please click here for the latest update of our data
Supported by NIGMS R01 GM59957.