Structure-based Ligand Discovery for GPCRs

G Protein Coupled Receptors (GPCRs) are the largest family of signaling proteins in the genome, sitting atop hundreds of transduction cascades that determine how cells and tissues respond to stimuli and to environmental cues. This, their direct accessibility to molecules outside the cell, and their great ligand binding sites, have made them the protein family most targeted by drugs.
The structural renaissance begun by the determination of the β2-adrenergic receptor has made ever-more of these receptors accessible to structure-based discovery. In proof-of-concept docking campaigns vs the β2, the A2a, D3, CXCR4, and muscarinic receptors, we observed hit rates ranging from 17% to over 50% (active hits/number experimentally tested), with potencies ranging from sub-nanomolar to low micromolar.
Bolstered by these results, we are seeking novel GPCR ligands that confer new biology. Three projects are being pursued.
1. With Bryan Roth’s group, we are deorphanizing the 120 orphan GPCRs in the genome. Combining empirical screening, modeling, and docking, specific probe molecules have emerged for three orphan GPCRs, including for GPR68, where the new probe (ogerin), a direct docking hit, was used to show that GPR68 was involved in fear-based learning in the mouse (Huang, 2015).
2. With the Sunahara, Kobilka, and Gmeiner labs we seek new allosteric molecules for GPCRs. Allosteric molecules have great signaling advantages, modulating the activities of primary transmitters without themselves signaling.
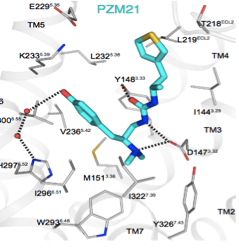
3. With the Roth, Gmeiner & Kobilka labs, we are seeking novel ligands for orthosteric sites for well-studied targets. Even against such sites, new chemotypes can confer new signaling properties, preferentially activating one GPCR effector over another. For instance, in the μ-opioid receptor, docking revealed a novel agonist that activated G protein signaling without activating arrestin signaling. In a mouse model, this new agonist (PZM21) conferred analgesia without respiratory depression and with reduced reinforcing behavior. As well as leading us into cool new biology in several areas, this project has become a testing ground for our latest docking and chemoinformatics methods.
Recent papers include:
- A Manglik, H Lin, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature (2016).
- XP Haung, J. Karpiak, et al. Allosteric ligands for the pharmacologically dark receptors GPR68 & GPR65. Nature 527, 477-83 (2015).
- S Wang et al., D4 dopamine receptor high-resolution structures enable the discovery of selective agonists.. Science 358, 381-386 (2017).
- K Lansu et al. In silico design of novel probes for the atypical opioid receptor MRGPRX2.. Nature Chem. Biol. 13, 529-536 (2017).
Supported by NIH U01104974, GM106990 and GM59957.